You should've already read the documentation before reading this.
Based on a preliminary analysis of the CASP6 results, the following observations can be made about the use of this server. Our group was one of the four server groups invited to give oral presentations at CASP6 (only the top performing groups are invited to give talks). We used the same automated servers for both the manual and automated predictions---the difference being that we were able to make a greater number of predictions for the latter due to relaxed time constraints. We recommend using the PROTINFO servers with these guidelines in mind.
The comparative modelling methods rely on the selection of an appropriate template (and an alignment for the former). The current default methods implemented for doing this seem to work best only on the easiest (clearly recognisable homology) cases. They usually fail on intermediate and the most difficult cases. To get around this problem, we recommend you first submit your sequence to the Bioinfo.pl metaserver and obtain the best result (alignment as well as template) from the 3D Jury system/method. The 3D Jury is a meta-predictor that combines the results of the other predictors and makes a consensus evaluation. This template choice (with all the main chain atoms present) as well as the alignment (for comparative modelling) can be submitted as input to the PROTINFO server. (Our main area of research is building loops and side chains accurately.)
The de novo method does indeed seem to consistently produce topologically accurate structures for small proteins (up to 100 residues) and/or fragments of a protein, even for the most difficult cases.
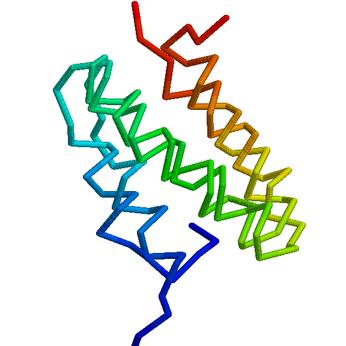
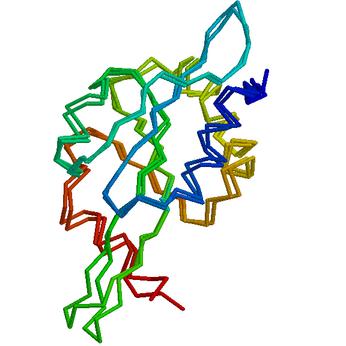
Here are some preliminary highlights (models are superposed on top of the target, and the direction of the CA trace is coloured) from the CASP6 experiment. Detailed evaluation of all our predictions is available at the CASP6 web site. All predictions shown below are our first models.
In addition, our comparative modelling module was evaluated as part of the LiveBench-7 and PDB-CAFASP-1 experiments (only the CM results are valid since the assessment is cumulative and our AB module cannot not make predictions for all targets). See for example the ranking of the method ``PRCM'' on easy and hard targets after selecting all servers in Livebench-7 (select ``plot added value'' to see another view); similarly for PDB-CAFASP-1. Note that the ROC results are not meaningful for our servers since the ``score'' we submitted was not reflective of model quality relative to experiment.
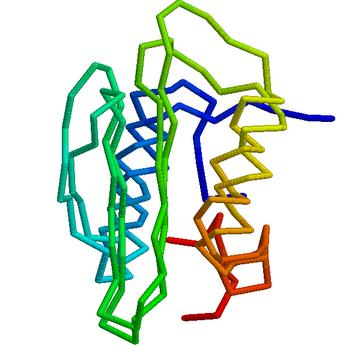
 PID to template: 80%
PID to template: 80%
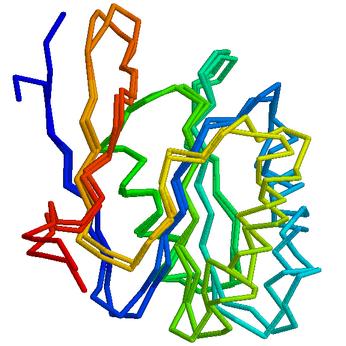 PID to template: 46%
PID to template: 46%